
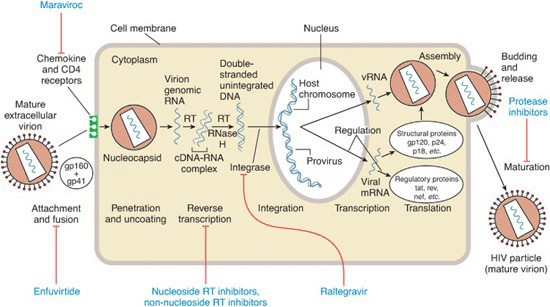
Pada kesempatan sebelumnya, telah dibahas mengenai klasifikasi serta mekanisme kerja dari obat-obatan antiretroviral atau ARV. Pada kesempatan kali ini kita akan mencoba membahas bagaimana profil obat ARV, khususnya ARV yang berada dan tersedia di Indonesia (Image by Christian Trick from Pixabay). Sebagai pembuka, kita tahu bahwa berdasarkan mekanisme kerjanya mempengaruhi siklus replikasi virus (gambar di bawah), ARV terbagi menjadi beberapa kelompok yaitu:
- Nucleoside/nucleotide reverse transcriptase inhibitor (NRTI/NtRTI)
- Non-nucleoside reverse transcriptase inhibitor (NNRTI)
- Protease inhibitor (PI)
- Entry inhibitor (EI)
- Integrase strand transfer inhibitor (INSTI)
Daftar Isi
Nucleoside/Nucleotide Reverse Transcriptase Inhibitor (NRTI/NtRTI)
Kelompok obat ini adalah salah satu obat pertama yang digunakan untuk pengobatan HIV. Sampai saat ini, NRTI menjadi tulang punggung bagi pengobatan lini pertama HIV. Sebagai kita ketahui, regimen HAART lini pertama terdiri dari setidaknya dua NRTI. Adapun beberapa obat yang termasuk ke dalam kelompok obat ini dan tersedia di Indonesia adalah zidovudin (AZT), lamivudine (3TC), tenofovir disoproxil (TDF), abacavir (ABC), dan emtricitabine (FTC). Berikut profil obat ARV golongan NRTI tersebut:

Zidovudine (AZT)
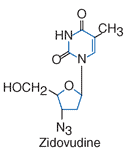
Zidovudine atau nama lainnya adalah azidothymidine (3′-azido-3′-deoxythymidine; AZT) merupakan salah satu ARV yang pertama kali digunakan untuk pengobatan HIV. Sama dengan kerja NRTI lainnya, zidovudine merupakan analog dari asam nukleat yang dalam hal ini adalah thymidine. Obat ini memiliki aktivitas antiretrovirus yang luas termasuk JIV-1, HIV-2, dan human T-cell lymphotrophic viruses (HTLV) I dan II. Adapun gugus kimia zidovudine adalah sebagai berikut:
Zidovudine akan berikatan dengan enzim reverse transcriptase dari HIV yang kemudian akan menyebabkan enzim tersebut berhenti bekerja. Secara selektif, zidovudine berikatan dengan enzim virus namun apabila dalam dosis besar, obat ini juga akan mengganggu enzim DNA polimerase dari manusia sehingga akan menimbulkan efek samping.
Zidovudine dapat diserap dari saluran cerna dengan cepat dan mencapai kadar puncak dalam darah dalam waktu 1 jam. Makanan dapat menurunkan kecepatan absorpsi namun kurva AUC tidak dipengaruhi sehingga boleh dikonsumsi bersamaan dengan makanan. Dosis umum zidovudine adalah 2 x 300 mg per hari. Obat ini dimetabolisme sebagian di hati (60-80% glucuronidase) sedangkan di ginjal hanya 14%. Pada keadaan gagal ginjal dimana eGFR <10, maka dilakukan perubahaan dosis menjadi 100 mg q6-8 jam. Tetapi, tidak dosis tidak diubah bagi penderita gagal ginjal yang menjalani hemodialisa.
Efek samping utama dari zidovudine adalah anemia dan granulositopenia sehingga pemantauan darah perifer lengkap diperlukan selama pasien mengonsumsi obat ini. Selain itu, efek samping lain berupa malaise, lemas, mual, dan muntah. Gejala ini biasanya akan menghilang setelah minggu pertama pemakaian.
Efek samping lain adalah perubahan warna pada kuku serta miopati dapat terjadi akibat penghambatan DNA mitokondria. Dapat pula terjadi hepatotoksisitas dan asidosis laktat namun jarang terjadi.
Lamivudine (3TC)
Lamivudine [(–)2′, 3′-dideoxy, 3′-thiacytidine; 3TC] merupakan analog dari cytidine. Obat ini aktif terhadap HIV-1, HIV-2, dan virus hepatitis B (HBV). Oleh sebab itu, obat ARV ini penting diberikan pada pasien HIV yang juga memiliki koinfeksi dengan hepatitis B. Obat ini memiliki afinitas yang rendah terhadap polimerase DNA manusia sehingga memiliki efek toksisitas yang rendah.

Sama dengan kelas analog lainnya, untuk menjadi aktif, lamivudine harus diubah menjadi bentuk molekul trifosfat di dalam sel. Proses fosforilasi lamivudine lebih efisien terjadi di sel yang dalam fase istirahat sehingga potensi lamivudine berkurang di sel mononuklear di perifer dibandingkan dengan pada galur sel (cell line).
Kombinasi lamivudine dengan zidovudine atau NRTI lainnya (kecuali emtricitabine karena efek hampir identik) memberikan efek yang menguntungkan. Virus yang mengalami resistensi terhadap lamivudine umumnya memiliki kemampuan replikasi yang rendah dan genome yang kurang stabil. Selain itu, mutasi yang menyebabkan resistensi terhadap lamivudine membuat virus tersebut kembali rentan atau tidak reisisten terhadap zidovudine dan tenofovir.
Lamivudine dapat diserap dari saluran cerna >80% dan tidak dipengaruhi makanan. Dosis lazim adalah 300 mg per hari bisa dijadikan 2 x 150 mg per hari. Lamivudine dimetabolisme <36% di hati dan sebagian besar (71%) disekresikan di ginjal. Oleh karena itu, harus dilakukan penyesuaian dosis apabila ada penurunan fungsi ginjal yaitu sebagai berikut:
- eGFR 30-49: 150 mg per hari
- eGFR 10-29: 150 mg per dosis pertama kemudian 100 mg per hari
- eGFR <10: 150 mg dosis pertama kemudian 50 mg per hari
- Menjalani hemodialisa: 50 mg dosis pertama kemudian 25 mg per hari
Efek samping lamivudine sangat jarang. Pemberian di atas dosis dapat menyebabkan neutropenia, nyeri kepala, dan mual muntah. Pernah terdapat laporan terjadi pankreatitis pada anak-anak namun hal ini tidak terkonfirmasi pada penelitian-penelitian lainnya.
Tenofovir Disoproxil (TDF)
Obat ARV ini adalah derivatif dari adenosine 5′-monophosphate tanpa cincin ribosa yang lengkap (gambar di bawah). Seperti lamivudine, obat ini aktif terhadap HIV-1, HIV-2, dan HBV. Tenofovir memiliki bioavailibiltas oral yang buruk dengan bentuk prodrug disoproxil diberikan agar memperbaiki bioavailibilitas dan daya penetrasi sel.

Bentuk trifosfat dari tenofovir merupakan bentuk aktif di dalam sel. Tenofovir memiliki afinitas rendah terhadap DNA polymerase-α, -β, and -γ sehingga tidak terlalu mempengaruhi mekanisme replikasi DNA manusia.
Tenofovir disoproxil hanya memiliki bioavailibilitas oral sebesar 25%. Makanan dengan kadar lemak yang tinggi dapat meningkatkan daya serap oral sampai 39%. Obat ARV ini tidak berikatan dengan protein plasma secara signifikan. T1/2 eliminasi dari obat ini adalah 14-17 jam. Tenofovir sebagian besar dieksresikan oleh ginjal baik melalui filtrasi maupun sekresi tubular aktif (70-80%) sedangkan metabolisme di hati sangat sedikit. Dosis umum tenofovir adalah 300 mg sekali sehari. Obat ini dosisnya harus dilakukan penyesuaian jika terjadi penurunan fungsi ginjal. Penyesuaian dosisnya adalah sebagai berikut:
- eGFR 30-49: 300 mg dua hari sekali (q48 jam)
- eGFR 10-29: 300 mg setiap 72-96 jam
- eGFR <10: 300 mg setiap 7 hari sekali
Efek samping tenofovir sebenarnya jarang dan secara umum pasien dapat menoleransi obat ini tanpa banyak masalah. Pada penelitian juga tidak didapatkan tenofovir dapat merusak sel tubular ginjal. Namun, terdapat laporan mengenai kasus jarang gagal ginjal akut dan sindrom Fanconi pada penggunaan tenofovir. Akibatnya, penggunaan obat ini harus dilakukan secara berhati-hati jika pasien juga mengalami gangguan fungsi ginjal.
Penggunaan tenofovir juga dilaporkan berhubungan dengan sedikit penurunan eGFR setelah penggunaan beberapa bulan. Dikarenakan perlunya dosis diubah seiring perubahan eGFR, maka fungsi ginjal pengguna tenofovir harus diperiksa secara berkala.
Emtricitabine (FTC)
Emtricitabine, (2R,5S)-5-fluoro-1-[2-(hydroxymethyl)-1,3-oxathiolan-5-yl] cytosine (FTC), merupakan analog dari cytidine dan secara kimiawi memiliki banyak kesamaan dengan lamivudine. Obat ARV ini aktif terhadap HIV-1, HIV-2, dan HBV.

Seperti halnya lamivudine, emtricitabine juga memiliki bentuk trifosfat sebagai bentuk aktif. Obat ini juga memiliki afinitas rendah terhadap polimerase DNA manusia sehingga efek samping atau toksisitasnya rendah. Penyebab resistensi emtricitabine juga mirip dengan lamivudine sehingga reaksi resistensi terhadap lamivudine juga berefek terhadap emtricitabine.
Obat ini diserap cepat oleh sistem pencernaan dan memiliki bioavailibilitas sampai 93%. Makanan dapat mengurangi Cmax tetapi secara umum AUV emtricitabine tidak dipengaruhi oleh makanan. Dibandingkan dengan analog nukleosida yang lain, emtricitabin memiliki waktu eliminassi yang lebih lambat dengan t1/2 8-12 jam. Metabolisme di hati hanya 13% sedangkan di ginjal 86%. Dosis umum emtricitabine adalah 200 mg per hari dengan penyesuaian dosis pada kondisi gangguan fungsi ginjal adalah sebagai berikut:
- eGFR 30-49: 200 mg dua hari sekali (q48 jam)
- eGFR 10-29: 200 mg setiap 72 jam
- eGFR <10: 200 mg setiap 96 jam
Obat ini memiliki sedikit sekali efek samping. Namun, penggunaan jangka panjang diketahui menyebabkan hiperpigmentasi pada kulit terutama pada daerah kulit yang banyak terpapar sinar matahari.
Abacavir (ABC)
Obat ARV ini merupakan analog purin karbosiklik sintetik dari golongan guanosine. Di bawah ini adalah gambar struktur molekul dari abacavir:

Abacavir diserap dengan baik oleh saluran cerna dan tanpa dipengaruhi oleh makanan dengan bioavailibilitas >80%. Obat ini dimetabolisme menjadi derivat asam 5′-karboksilat yang dikatalisasi oleh alkohol dehidrogenase dan menjadi 5′-glukurinida oleh proses glukuronidasi. Metabolit ini meliputi 30% dan 36% dari total proses eliminasi abacavir. Obat ini bukan merupakan substrat atau inhibitor CYP dan 50% terikat dengan protein plasma. Hanya <5% dari obat ini dieliminasi oleh ginjal. Dosis lazim adalah 600 mg per hari dan tidak diperlukan penyesuaian dosis pada penurunan fungsi ginjal.
Abacavir memiliki efek samping yang unik namun fatal yaitu sindrom hipersensitivitas. Ciri dari sindrom ini adalah demam, nyeri perut, dan gangguan saluran cerna lainnya. Selain itu disertai pula dengan rash makulopapular ringan dan keluhan lelah. Keluhan lain dapat berupa gangguan pernapasan, nyeri otot, nyeri kepala, dan parestesia. Keluhan tersebut muncul dalam 6 minggu sejak mulai pemberian obat abacavir. Jika hal ini terjadi, maka abacavir harus dihentikan dan pasien tidak dapat lagi diberikan abacavir. Selain sindrom hipersensitivitas ini, abacavir jarang memberikan efek samping lain.
Non-Nucleoside Reverse Transcriptase Inhibitor (NNRTI)
Profil obat ARV selanjutnya adalah dari golongan NNRTI. Golongan obat ini menghambat enzim reverse transcriptase dengan menempel pada kantung hidrofobik pada subunit p66 dari enzim HIV tersebut. Tempat kantung ini tidak secara langsung menghentikan enzim dan jauh dari tempat aktif dari enzim. Namun, obat ini merupah konformitas enzim sehingga kerja enzim akan menjadi sangat berkurang. Beda dengan NRTI, NNRTI tidak memerlukan proses fosforilisasi untuk menjadi aktif. Obat ini juga tidak mengganggu polimerase DNA dari manusia. Dua obat yang banyak digunakan dari glongan ini adalah efavirenz dan nevirapine.

Semua obat golongan ini dimetabolisme oleh hati. Nevirapine adalah substrat dari CYP3A4 sedangkan efavirenz adalah substrat dari CYP2B6 dan CYP3A4.
Efavirenz (EFV)
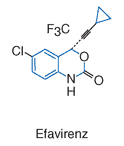
Efavirenz adalah NNRTI dengan nama kimia 1,4-dihydro-2H-3,1-benzoxazin-2-one. Sama dengan NNRTI lain, efavirenz hanya aktif terhadap HIV-1.
Obat ini diabsorpsi dengan baik oleh saluran cerna dan mencapai kadar puncak di plasma dalam 5 jam. Terjadi penurunan absoprsi ketika dosis obat dinaikan dan bioavailibilitas naik 22% saat obat diminum bersamaan dengan makanan uang kaya lemak. Efavirenz berikatan dengan plasma >99%. Biasanya obat ini harus diminum saat perut kosong sebelum tidur untuk mengurangi efek samping.
Efavirenz dieliminasi melalui metabolisme oksidasi terutama oleh CYP2B6 dan sedikit oleh CYP3A4. Obat utama tidak diekskresikan secara signifikan oleh ginjal dan memerlukan t1/2 40-55 jam. Dosis umum dari obat ARV ini adalah 600 mg per hari.
Efek samping utama terutama akibat toksisitas terhadap susunan saraf pusat (SSP) dan efek psikiatri. TErjadi pada 53% pasien namun hanya <5% pasien dihentikan pengobatannya karena efek samping ini. Efek samping SSP ini terjadi pada dosis pertama dan dapat bertahan beberapa jam. Beberapa gejala yang lebih berat bahkan menghilang setelah beberapa minggu. Gejala yang dikeluhkan biasanya pusing, gangguan konsenterasi, disforia, mimpi buruk, dan insomnia. Bahkan dapat pula terjadi gejala psikosis. Untungnya, gejala psikiatri ini dapat berkurang dalam 4 minggu setelah pemakaian.
Efek samping lainnya adalah rash yang dapat terjadi pada 27% kasus namun hilang setelah beberapa sendiri dan jarang menimbulkan masalah. Sindrom Steven-Johnson pernah dilaporkan namun jarang terjadi. Selain itu, efavirenz dapat pula menyebabkan peningkatan transaminase, dan kolesterol.
Obat ini adalah ARV satu-satunya yang bersifat teratogenik pada primata. Namun, data untuk kejadian teratogenik pada manusia kurang mendukung efek teratogenik efavirenz pada janin.
Nevirapine (NVP)
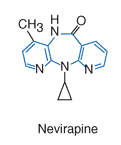
Nevirapine adalah NNRTI dari henis dipyridodiazepinone dengan aktivitas yang poten terhadap HIV-1. Obat ini tidak memiliki aktivitas terhadap HIV-2 ataupun jenis retrovirus lainnya.
Obat ARV ini dapat diserap dengan baik oleh saluran cerna dan tidak dipengaruhi makanan ataupun antasida. Obat ini juga dapat melewati plasenta dan terdapat di ASI sehingga banyak disarankan untuk dipakai dalam mencegah transmisi HIV dari ibu ke bayi.
Rute eliminasi dari nevirapine adalah dengan metabolisme oksidatif yang melibatkan CYP3A4 dan CYP2B6. Hanya kurang dari 3% dari obat ini dibuang di urin tanpa diubah terlebih dahulu. Waktu eliminasi t1/2 dari obat ini cukup panjang yaitu 25-30 jam dan lebih lama lagi pada individu tertentu khususnya keturunan Afrika.
Selain itu, obat ini juga merupakan inducer bagi enzim CYP3A4 sehingga mempengaruhi kecepatan eliminasi dirinya sendiri. Saat pertama kali dikonsumsi, t1/2 dari nevirapine adalah 45 jam dan kemudian berkurang menjadi 25-30 jam setelah 2 minggu pemakaian. Hal tersebut menyebabkan obat ini disarankan diberikan pada dosis 200 mg sekali sehari pada 14 hari pertama kemudian baru dinaikan 2 x 200 mg apabila tidak ada masalah. Beberapa penelitian juga melihat bahwa dosis 200 mg per hari sudah cukup walaupun hal ini belum atau tidak diadopsi sebagai dosis standar.
Efek samping yang paling sering ditemukan adalah rash sekitar 16%. Erupsi makular atau papular ringan biasanya meliputi badan, wajah, dan ekstremitas dan muncul pada 6 minggu pertama konsumsi obat. Gatal juga kadang dikeluhkan. Pada kebanyakan pasien, gejala ini dapat hilang seiring dengan diteruskannya pemakaian obat. Pemberian glukokortikoid dapat memperburuk kondisi rash. Sindrom Steven-Johnson dapat terjadi walaupun jarang.
Peningkatan enzim transaminase terjadi pada sampai 14% pengguna. Hepatitis imbas obat muncul pada 1% pasien. Kejadian hepatitis yang berat dan fatal dapat muncul khususnya pada wanita dengan CD4 ≥250 sel/mm3 khususnya saat hamil. Efek samping lainnya termasuk demam, lelah, nyeri kepala, somnolen, dan mual.
Dosis umum efavirenz adalah 600 mg per hari walaupun dapat diturunkan menjadi 400 mg per hari. Dikarenakan ekskresi oleh ginjal yang kecil, tidak diperlukan penyesuaian dosis efavirenz apabila terjadi penurunan fungsi ginjal.
Protease Inhibitor (PI)
Obat ARV selanjutnya adalah protease inhibitor (PI). Golongan obat ini juga merupakan salah satu anggota penting dari HAART. Di negara kita, PI terutama khususnya lovinapir adalah bagian dari lini kedua pengobatan HIV.
Kelompok obat ini terdiri dari molekul mirip peptida yang menghambat kerja dari aspartyl protease dari virus. Protease ini terdiri dari dua monomer yang masing-masing terdiri dari rangkaian 99 asam amino. Setiap monomer menyumbangkan residu asam aspartat sebagai katalis untuk reaksi proteasae. Tempat pembelahan dari enzim ini adalah sisi N terminal dari residu prolin, terutama antara fenilalanin dan prolin. Adapun aspartyl protease dari manusia (renin, pepsin, gastricsin, dan cathepsins D dan E) hanya memiliki satu rantai polipeptida sehingga tidak dihambat secara signifikan oleh inhibitor terhadap protease HIV.
Obat golongan PI ini menghambat pembelahan prekursor gag dan pol baik itu struktur virus maupun protein enzimatik dari virus. Hal ini menyebabkan pencegahan metamorfosis partikel virus menjadi virus yang matur. Di bawah ini adalah bagan mengenai molekul PI dan bagaimana tempat obat tersebut bekerja.

Salah satu ciri dari kelompok obat ini adalah tingginya variabilitas antar individu yang menggambarkan perbedaan aktivitas CYP hati maupun usus antar individu. Proses eliliminasi atau clearence terutama melalui proses metabolisme di hati. Semua agen kecuali nelfinavir dimetabolisme oleh CYP3A4. Kebanyakan PI juga terikat secara kuat pada protein plasma.
Oleh karena dimetabolisme oleh CYP4A4, maka salah satu efek samping penting dari PI adalah potensi interaksi dengan berbagai macam obat. Kebanyakan PI menghambat kerja CYP3A4 terutama ritonavir yang merupakan inhibitor terkuat. Adalah hal yang umum dengan menggabungkan obat PI dengan ritonavir dosis rendah (ritonavir boosted, /r) untuk memanfaatkan kemampuan ritonavir menghambat metabolisme obat utama sehingga memperlambat eliminasi obat.
Ritonavir (RTV atau /r)
Obat ARV ini adalah inhibitor protease HIV peptidomimetik yang mencoba mengikuti struktur komplemen simetri dari aksis C2 situs aktif protease virus. Obat ini aktif terhadap HIV-1 dan HIV-2 walaupun sedikit kurang aktif terhadap HIV-2.

Ritonavir digunakan terutama sebagai enhancer atau penguat dosis obat lain. Jika ditujukan untuk keperluan tersebut, maka dosis yang diberikan adalah dosis kecil. Tidak diketahui bagaimana pengaruh praktek pemberian dosis rendah ritonavir ini dapat mempengaruhi resistensi HIV terhadap ritonavir.
Absoprsi dari ritonavir berlangsung cepat dan sedikit dipengaruhi oleh makanan atau pun formulasi. Variabilitas antar individu untuk obat ini tinggi dengan tingkat variasi konsenterasi obat dapat mencapai 6 kali lipat pada individu yang mendapat ritonavir 600 mg setiap 12 jam.
Metabolisme obat ini seperti disebutkan sebelumnya teritama adalah oleh CYP3A4 dan sebagian ekcil oleh CYP2D6. Rute eliminasae ritonavir dan metabolitnya adalah terutama melalui feses (86%). Hanya 3% saja yang dibuang melalui urin. Obat ini menginduksi metabolisme dirinya sendiri dan tercapai kondisi steady state setelah pemberian obat 2 minggu. Ritonavir terikat pada protein plasma sebesar 98-99%.
Efek samping yang utama adalah gejala mual, muntah, diare, anoreksia, nyeri perut, dan perubahan perasa. Efek ini dapat berkurang apabila obat diminum bersamaan dengan makanan. Parestesia perifer atau perioral dapat terjadi pada pemberian dosis 2 x 600 mg. Efek samping ini akan berkurang setelah beberapa minggu terapi. Obat ini juga dapat menyebabkan peningkatan kadar kolesterol dan trigliserida dari plasma yang juga disertai dengan tanda lipodistrofi dan mungkin meningkatkan proses aterosklerosis pada beberapa pasien.
Patut dicatat juga efek ritonavir terhadap obat lain. Dikarenakan menghambat CYP3A4, maka dapat meningkatkan konsenterasi obat lain yang dimetabolisme oleh enzim ini. Efek inhibisi ritonavir bersifat reversibel dan efeknya dapat bertahan setalah 2-3 hari pemberian ritonavir dihentikan. Rifampicin yang merupakan induser kuat CYP3A4 dapat mengurangi dosis darah ritonavir sehingga perlu penyesuaian dosis obat.
Untuk tujuan sebagai booster obat lain, dosis ritonavir yang diperlukan adalah 100 mg atau 200 mg perhari. Sedangkan untuk tujuan terapetik, dosis yang diberikan adalah 2 x 600 mg per hari.
Lopinavir (LVP)
Lovinapir merupakan peptidomimetik yang mirip dengan ritonavir namun memiliki potensi 3-10 kali lebih poten terhadap HIV-1 secara in vitro dibanding dengan ritonavir. Selain terhadap HIV-1, lovinapir juga aktif terhadap HIV-2. Saat ini hanya tersedia sediaan koformulasi lovinapir dengan ritonavir dosis rendah (LVP/r).

Obat ini secara cepat diserap oleh saluran cerna dan proses penyerapannya tidak dipengaruhi oleh makanan. Pada koformulasi, rasio lovinapir dengan ritonavir biasanya adalah 4:1 namun konsentrasi ini berubag dalam darah menjadi 20:1. Hal ini disebabkan konsentrasi lovinapir dalam darah ditingkatkan akibat hambatan ritonavir terhadap CYP3A4. Sekitar 90% metabolisme lovinapir dilakukan oleh oksidasi oleh CYP3A4 dan hanya <3% saja yang dikeluarkan di urin.
Dosis 50 mg dari ritonavir dapat meningkatkan AUC lovinapir sampai 77 kali dibandingkan jika 400 mg lovinapir diberikan secara sendiri. Sedangkan 100 mg ritonavir meningkatkan AUC sampai 155 kali. Dosis umum dari lovinapir adalah 2 x 200 mg yang dikombinasi dengan ritonavir 2 x 50-100 mg per hari.
Efek samping yang didapat pada pemberian LVP/r adalah gangguan saluran cerna berupa diare, mual, dan muntah. Efek samping ini lebih ringan dibandingkan dengan dosis 2 x 600 mg tanpa ritonavir. Sedangkan gangguan laboratorium yang dijumpai adalah peningkatan kolesterol dan trigliserida.
Atazanavir
Obat ini merupakan protease inhibitor azapeptida dengan struktur kimia C2 simetris yang aktif baik terhadap HIV-1 maupun HIV-2. Atazanavir diserap di saluran cerna secara cepat dengan kadar puncak tercapai 2 jam setelah diminum.

Absorvsi di saluran cerna dipengaruhi oleh makanan dimana cemilan meningkatkan AUC 70% sedangkan makanan tinggi lemak meningkatkan AUC 35%. Oleh sebab itu obat ini disarankan diminum bersamaan dengan waktu makan. Absorpsi atazanavir berkurang apabila keasaman lambung menurun sehingga obat-obatan seperti PPI mengurangi penyerapan obat ini.
Metabolisme oksidatif azatanavir utamanya dilakukan oleh hati terutama oleh CYP3A4. Hanya 7% porsi obat yang dikeluarkan tanpa diubah melalui urin. Rerata eliminasi t1/2 pada dosis standar 400 mg adalah 7 jam namun pola eliminasi ini non linier dengan t1/2 pada dosis 600 mg adalah 10 jam.
Atazanavir dalam plasma berikatan dengan protein plasma baik albumin (86%) maupun asam α1-glikoprotein. Pada cairan serebrospinal, konsenterasi obat hanya mencapai <3% dari konsenterasi plasma.
Efek samping obat ini adalah terjadinya hiperbilirubinemia tanpa terkonjugasi namun dengan mekanisme yang tidak melibatkan hati (non hepatotoksik). Hal ini disebabkan karena obat ini menghambat UDP-glucuronosyl transferase. Efek lain adalah munculnya batu empedu.
Efek samping lain adalah diarea dan mual terutama pada beberapa minggu pertama pemakaian. Dibandingkan dengan lopinavir, obat ini menimbulkan kadar kolesterol dan trigliserida yang lebih rendah.
Entry Inhibitor (EI)
Golongan obat ini menghambat proses masuknya virus ke dalam sel CD4. Terdapat dua obat pada kelompok ini dengan mekanisme yang berbeda yaitu maraviroc dan enfuvirtide. Sayangnya, kedua obat ini belum banyak beredar di Indonesia.
Maravirov (MVC)
Maraviroc merupakan antaginis reseptor kemokin CCR5 mengikat gp120. Obat ARV ini otomatis hanya aktif terhadap HIV yang memiliki troposme terhadap CCR5 dan tidak efektif terhadap virus dengan tropisme terhadap CXCR4 atau keduanya (dual tropism).

Bioavailibilitas dari maraviroc adalah 23-33% dan tergantung dari dosis yang diberikan. Makanan dapat mengurangi AUC sampai 50% sehingga disarankan meminum maraviroc dalam keadaan perut kosong. Obat ini mengikat protein plasma sekitar 76% dan eliminasi terutama melalui metabolisme oleh CYP3A4 dengan waktu t1/2 eliminasii 10,6 jam.
Maraviroc secara umum dapat ditoleransi dengan baik dengan reaksi toksisitas yang minimal. Pernah dilaporkan satu kasus heptotoksisitas yang serius namun dalam pengamatan, jumlah hepatotoksisitas ini tidak jauh beda dengan plasebo. Jika digabungkan dengan obat inhibitor CYP3A4, maka dosis awal adalah 2 x 150 mg sedangkan jika digabungkan dengan obat induser CYP3A4 maka dosis awal adalah 2 x 600 mg. Untuk konkomitan dengan obat selain itu, dosis awal adalah 2 x 300 mg.
Enfuvirtide (ENF)
Obat ini adalah peptida sintetik yang terdiri atas 36 asam amino yang sekuensinya berasal dari bagian transmembran gp41. Bagian ini terlibat dalam fusi membran virus dengan membran sel. Enfuvirtide tidak aktif terhadap HIV-2.
Awalnya obat ini ditujukan sebagai komponen vaksin karena memiliki derajat konservasi tinggi di berbagai strain HIV-1. Namun, ternyata pada tes, enfuvirtide juga aktivitas anti HIV yang poten. Dikarenakan bentuknya yang besar, obat ini mahal untuk dibuat dan diberikan secara subkutan sehingga tidak praktis.

Enfuvirtide memiliki mekanisme unik. Peptida ini dapat memblok interaksi antara sekuens N36 dan C34 dari glikoprotein gp41. Caranya adalah dengan mengikat bagian hidrofobik dari koil N36. Hal ini mencegah pembentukan bundel 6 heliks yang penting untuk proses fusi membran dan masuknya virus ke dalam sel. Obat ini efektif terhadap virus yang resisten terhadap obat lainnya. Walaupun demikian, virus yang membawa mutasi pada gen yang mengkodekan gp41 dapat menjadi kebal terhadap obat ini.
Obat ini diberikan secara parenteral yaitu subkutan. Metode eliminasi belum diketahui secara pasti dengan lama t1/2 sekitar 3,8 jam sehingga harus diberikan setiap 2 kali sehari. Sebagian besar efek samping yang terjadi adalah efek samping lokal di sekitar tempat injeksi. Efek samping tersebut bisa nyeri, kemerahan atau eritema, dan indurasi.
Integrase Strand Transfer Inhibitor (INSTI)
Obat ARV golongan INSTI jarang sekali ditemukan di Indonesia. Walaupun demikian, obat ini adalah salah satu obat penting baik pada penatalaksanaan infeksi HIV. INSTI berfungsi mencegah integrasi genome virus dengan kromosom sel inang. Mekanismenya adalah dengan mengikat enzim integrase yang berperan penting pada proses integrasi ini.

Raltegravir adalah anggota dari kelompok INSTI. Obat ini aktif terhadap HIV-1 dan HIV-2. Bioavailibilitas oral cukup bervariasi dan dapat mencapai konsenterasi puncak di darah dalam waktu 1-3 jam. Dosis lazimnya adalah 2 x 400 mg.
Obat ini secara umum dapat ditoleransi pasien dengan baik dengan sedikit efek samping. Raltegravir dimetabolisme oleh UGT1A1 sehingga dapat berinteraksi dengan obat lain yang memiliki jalur metabolisme yang sama. Obat yang dapat meningkatkan konsenterasi raltegravir contohnya adalah atazanavir sedangkan yang dapat menurunkan contohnya adalah tenofovir. Ketika raltegravir digabung dengan rifampicin, maka dosis raltegravir harus ditingkatkan menjadi 2 x 800 mg per hari.
Kesimpulan
Walaupun saat ini perkembangan obat-obatan ARV sudah demikian maju namun tetap ditemukan beberapa masalah. Hal yang paling menjanjikan adalah bahwa saat ini ARV dapat dijangkau oleh penderita di negara berkembang maupun negara dunia ketiga. Namun, tentu tidak semua jenis ARV dan dengan makin berkembangnya perkembangan resistensi, ketersediaan obat-obatan ARV yang lengkap sangat diperlukan terutama untuk menangani kasus-kasus resisten.
Pada akhirnya, kepatuhan pasien menjalani terapi menjadi sangat penting. Tanpa kepatuhan minum obat yang baik, resistensi dapat berkembang dengan cepat. Keterbatasan penelitian dan sulitnya mengembangkan obat menjadi faktor yang benar-benar harus dipertimbangkan dan diedukasikan kepada pasien untuk bekerjasama dalam menangani dan mencegah terjadinya resistensi pada obat-obatan ARV ini.
Sumber
- de Béthune M-P. Non-nucleoside reverse transcriptase inhibitors (NNRTIs), their discovery, development, and use in the treatment of HIV-1 infection: A review of the last 20 years (1989–2009). Antiviral Res. 2010 Jan;85(1):75–90.
- Eckhardt BJ, Gulick ROYM. Infectious Diseases. 4th ed. Cohen J, Powderly WG, Opal SM, editors. Infectious Disease. Elsevier; 2017. 1292-308 p.
- Flexner C. Antiretroviral agents and treatment of HIV infection. In: Brunton LL, Chabner BA, Knollmann BC, editors. Goodman & Gilman’s the pharmacological basis of therapeutics. 12th ed. New York: McGraw-Hill; 2011. p. 1451–82.
- Hammer SM, Squires KE, Hughes MD, Grimes JM, Demeter LM, Currier JS, et al. A Controlled Trial of Two Nucleoside Analogues plus Indinavir in Persons with Human Immunodeficiency Virus Infection and CD4 Cell Counts of 200 per Cubic Millimeter or Less. N Engl J Med. 1997 Sep 11;337(11):725–33.
- World Health Orzanization. Consolidated guidelines on the use of antiretroviral drugs for treating and preventing HIV infection: Recommendations for a public health approach. 2nd ed. Geneva: WHO; 2016.

Seorang dokter, saat ini sedang menjalani pendidikan dokter spesialis penyakit dalam FKUI. Peminat berbagai topik sejarah dan astronomi.
Comments 1
terima kasih kaka dokter